


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
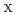 (X = 1, 2)
(X = 1, 2)Kido, Kentaro
International Journal of Quantum Chemistry, 121(21), p.e26781_1 - e26781_15, 2021/11
Times Cited Count:1 Percentile:16.75(Chemistry, Physical)Miyahara, Naoya; Miwa, Shuhei; Horiguchi, Naoki; Sato, Isamu*; Osaka, Masahiko
Journal of Nuclear Science and Technology, 56(2), p.228 - 240, 2019/02
Times Cited Count:7 Percentile:61.18(Nuclear Science & Technology)In order to improve LWR source term under severe accident conditions, the first version of a fission product (FP) chemistry database named "ECUME" was developed. The ECUME is intended to include major chemical reactions and their effective kinetic constants for representative SA sequences. It is expected that the ECUME can serve as a fundamental basis from which FP chemical models in the SA analysis codes can be elaborated. The implemented chemical reactions in the first version were those for representative gas species in Cs-I-B-Mo-O-H system. The chemical reaction kinetic constants were evaluated from either literature data or calculated values using ab-initio calculations. The sample chemical reaction calculation using the presently constructed dataset showed meaningful kinetics effects at 1000 K. Comparison of the chemical equilibrium compositions by using the dataset with those by chemical equilibrium calculations has shown rather good consistency for the representative Cs-I-B-Mo-O-H species. From these results, it was concluded that the present dataset should be useful to evaluate FP chemistry in Cs-I-B-Mo-O-H system under LWA SA conditions.
 system
systemKurosaki, Yuzuru; Takayanagi, Toshiyuki*
Chemical Physics Letters, 406(1-3), p.121 - 125, 2005/04
no abstracts in English
Kurosaki, Yuzuru; Yokoyama, Keiichi; Teranishi, Yoshiaki
Chemical Physics, 308(3), p.325 - 334, 2005/01
Times Cited Count:23 Percentile:60.42(Chemistry, Physical)A total of  1200 trajectories have been integrated for the two dissociation channels of formic acid, HCOOH
1200 trajectories have been integrated for the two dissociation channels of formic acid, HCOOH  HO + CO (1) and HCOOH CO + H (2), which occur with 248 and 193 nm photons, using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. It was found that the percentage of the energy distributed to a relative translational mode in reaction 2 is much larger than that in reaction 1. This is mainly due to the difference in the geometry of transition state (TS); the HO geometry in the TS of reaction 1 was predicted to significantly deviate from the equilibrium one, whereas the CO and H geometries in the TS of reaction 2 were found to be more similar to their equilibrium ones. It was also found that the product diatomic molecules, CO and H, are both vibrationally and rotationally excited. The calculated relative population of the vibrationally excited CO for the 248 nm photodissociation was consistent with experiment.
HO + CO (1) and HCOOH CO + H (2), which occur with 248 and 193 nm photons, using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. It was found that the percentage of the energy distributed to a relative translational mode in reaction 2 is much larger than that in reaction 1. This is mainly due to the difference in the geometry of transition state (TS); the HO geometry in the TS of reaction 1 was predicted to significantly deviate from the equilibrium one, whereas the CO and H geometries in the TS of reaction 2 were found to be more similar to their equilibrium ones. It was also found that the product diatomic molecules, CO and H, are both vibrationally and rotationally excited. The calculated relative population of the vibrationally excited CO for the 248 nm photodissociation was consistent with experiment.
 potential energy surfaces for the lowest three doublet states (1
potential energy surfaces for the lowest three doublet states (1 A', 2A', and 1A") of the BrH system
A', 2A', and 1A") of the BrH systemKurosaki, Yuzuru; Takayanagi, Toshiyuki
Journal of Chemical Physics, 119(15), p.7838 - 7856, 2003/10
Times Cited Count:26 Percentile:63.62(Chemistry, Physical)Adiabatic potential energy surfaces of the lowest three doublet states (1A', 2A', and 1A") for the BrH system have been calculated globally using the MRCI+Q method with the aug-cc-pVTZ basis set. Spin-orbit effects were considered on the basis of Breit-Pauli Hamiltonian. The calculated adiabatic energies were fitted to the analytical functional form of many-body expansion. The barrier heights of the abstraction and exchange reactions on the ground-state PES were calculated to be 1.28 and 11.71 kcal mol , respectively, at the MRCI+Q/aug-cc-pVTZ level of theory. The fits for the three PESs were successful within the accuracy of 0.1 kcal mol. Thermal rate constants for the abstraction and exchange reactions and their isotopic variants were calculated with the fitted 1A' PES using the ICVT/LAG method. The calculated rate constants for the abstarction reactions agree fairly well with experiment but those for the exchange reactions were much smaller than experiment, which suggests that the reliable experimental data are still insufficient.
, respectively, at the MRCI+Q/aug-cc-pVTZ level of theory. The fits for the three PESs were successful within the accuracy of 0.1 kcal mol. Thermal rate constants for the abstraction and exchange reactions and their isotopic variants were calculated with the fitted 1A' PES using the ICVT/LAG method. The calculated rate constants for the abstarction reactions agree fairly well with experiment but those for the exchange reactions were much smaller than experiment, which suggests that the reliable experimental data are still insufficient.
Miyazaki, Mikiya*
JAERI-Data/Code 2003-007, 55 Pages, 2003/05
 JAERI-Data-Code-2003-007.pdf:2.53MB
JAERI-Data-Code-2003-007.pdf:2.53MB
Generally it is well known that the R&D works on new materials or devices will play a central role on the evolution of future society. The structure of a substance and material is needed to be dealt with in detail by quantum mechanics, because the limit on accuracy has come in sight in the calculation using a classical theory. The research on the latest electronic state calculation technique founded on quantum mechanics made a great advance as the technique of solving these problems as well as the technique of a computational materials design. It enables the prediction of material properties because it is based on First Principles. Therefore, in the future it is expected to have a very high possibility of becoming a breakthrough in such a situation. In this article, the example calculation results by PC cluster on the codes used in the Computational Materials Design workshop, held on Sep.19-21, at ITBL-Building and IIAS under the auspices of the University of Osaka, are described. Furthermore, the graphical user interfaces on the codes are examined.
 CHOCH+HCO; Direct ab initio molecular dynamics study
CHOCH+HCO; Direct ab initio molecular dynamics studyKurosaki, Yuzuru; Yokoyama, Keiichi
Chemical Physics Letters, 371(5-6), p.568 - 575, 2003/04
Times Cited Count:31 Percentile:69.39(Chemistry, Physical)A total of 400 trajectories for the photodissociation, CHCHOCH+HCO, on the T potential surface have been calculated using the direct ab initio molecular dynamics method at the UB3LYP/cc-pVDZ level of theory. It was predicted that the product CH is neither vibrationally nor rotationally excited and HCO is vibrationally not excited but rotationally excited. The averaged HCO rotational energy was calculated to be 1.1 kcal/mol, which is 15.1 % of the available energy, 7.3 kcal/mol. The present result agrees with experiment within just a few percent of the observed data.
potential surface have been calculated using the direct ab initio molecular dynamics method at the UB3LYP/cc-pVDZ level of theory. It was predicted that the product CH is neither vibrationally nor rotationally excited and HCO is vibrationally not excited but rotationally excited. The averaged HCO rotational energy was calculated to be 1.1 kcal/mol, which is 15.1 % of the available energy, 7.3 kcal/mol. The present result agrees with experiment within just a few percent of the observed data.
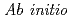 study of hydrogen hydrate clathrates for hydrogen storage within the ITBL environment
study of hydrogen hydrate clathrates for hydrogen storage within the ITBL environmentSluiter, M. H. F.*; Belosludov, R. V.*; Jain, A.*; Belosludov, V. R.*; Adachi, Hitoshi*; Kawazoe, Yoshiyuki*; Higuchi, Kenji; Otani, Takayuki
Lecture Notes in Computer Science 2858, p.330 - 341, 2003/00
Recently, for the first time a hydrate clathrate was discovered with hydrogen. Aside from the great technological promise that is inherent in storing hydorogen at high density at modest pressures, there is great scientefic interest as this would constitute the first hydrate clathrate with multiple guest molecules per cage. The multiple cage occupancy is controversial, and reproducibility of the experiments has been questioned. Therefore in this study we try to illucidate the remarkable stability of the hydrogen hydrate clathrate, and determine the thermodynamically most favored cage occpancy using highly accrate computer simulations in a parameter survey. To carry out these extraordinary demanding computations a distributed code has been developed using the SuperSINET with the ITBL software as the top-layer.
CHOCH +CO: Direct ab initio dynamics study
+CO: Direct ab initio dynamics studyKurosaki, Yuzuru; Yokoyama, Keiichi
Journal of Physical Chemistry A, 106(47), p.11415 - 11421, 2002/11
Times Cited Count:36 Percentile:72.97(Chemistry, Physical)A total of 100 trajectories for the photodissociation, CHCHO CH + CO, on the S0 potential surface have been calculated using the direct ab initio molecular dynamics method at the RMP2(full)/cc-pVDZ level of theory. The energy distributions for the relative translational energy, the CO internal energy, and the CH internal energy were calculated to be 28, 20, and 51 %, respectively. It was predicted that the product CO is highly rotationally excited but vibrationally almost not excited; on average, the rotational and vibrational quantum numbers were 68.2 and 0.15, respectively, which qualitatively agrees with the recent observation of Gherman et al. (J. Chem. Phys. 2001, 114, 6128.)
H + Cl CHCl + Cl reaction; Ab initio molecular orbital studyKurosaki, Yuzuru
Journal of Molecular Structure; THEOCHEM, 545(1-3), p.225 - 232, 2001/07
The CASSCF and MRCI calculations with the cc-pVTZ basis set have been carried out for the CH + Cl CHCl + Cl reaction. It has been revealed that the reaction has a small barrier from the CHCl + Cl side at the CASSCF level of theory, but it has no barrier at the MRCI level. Namely, the CHCl + Cl CH + Cl reaction was predicted to be a spontaneous reaction. The result of the MRCI calculation strongly supports the prediction of our previous PMP4(SDTQ) calculation [J. Mol. Struct. (Theochem) 503 (2000) 231].
at 157.6nm, 2; A Theoretical study using wave packet propagationYokoyama, Keiichi; Yokoyama, Atsushi; Takayanagi, Toshiyuki
Journal of Chemical Physics, 114(4), p.1624 - 1630, 2001/01
Times Cited Count:4 Percentile:12.22(Chemistry, Physical)no abstracts in English
Nakazawa, Tetsuya; Yokoyama, Keiichi; Grismanous, J.*; Katano, Yoshio*
Journal of Nuclear Materials, 279(2-3), p.201 - 206, 2000/06
Times Cited Count:10 Percentile:57.02(Materials Science, Multidisciplinary)no abstracts in English
 A' electronic states of H
A' electronic states of H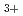
Ichihara, Akira; Yokoyama, Keiichi; Iwamoto, Osamu
JAERI-Data/Code 98-031, 18 Pages, 1998/11
JAERI-Data-Code-98-031.pdf:0.89MB
no abstracts in English
Ikezoe, Yasumasa; Suzuki, Kazuya; Nakashima, Mikio; Yokoyama, Atsushi; Shiraishi, Hirotsugu; Ono, Shinichi*
JAERI-Research 98-051, 43 Pages, 1998/09
JAERI-Research-98-051.pdf:1.63MB
no abstracts in English
; Yokoyama, Keiichi; Noda, Kenji
Journal of Nuclear Materials, 258-263, p.571 - 575, 1998/00
Times Cited Count:14 Percentile:72.98(Materials Science, Multidisciplinary)no abstracts in English
loss reaction from ethane cation, CH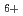
Kurosaki, Yuzuru*; Takayanagi, Toshiyuki
Chemical Physics Letters, 277(4), p.291 - 298, 1997/00
Times Cited Count:10 Percentile:35.5(Chemistry, Physical)no abstracts in English
system with the trajectory-surface-hopping modelIchihara, Akira; Shirai, Toshizo; Yokoyama, Keiichi
Journal of Chemical Physics, 105(5), p.1857 - 1861, 1996/08
Times Cited Count:38 Percentile:77.95(Chemistry, Physical)no abstracts in English
S+O( P)HS+OH reaction; A Theoretical study
P)HS+OH reaction; A Theoretical studyYokoyama, Keiichi; Takayanagi, Toshiyuki
Journal of Chemical Physics, 104(5), p.1953 - 1957, 1996/02
Times Cited Count:5 Percentile:21.07(Chemistry, Physical)no abstracts in English
CN molecule; An Experimental and ab initio studyHashimoto, Masashi; Yokoyama, Keiichi; Kudo, Hiroshi*; Wu, C. H.*; P.von-R.Schleyer*
Journal of Physical Chemistry, 100(39), p.15770 - 15773, 1996/00
Times Cited Count:13 Percentile:43.76(Chemistry, Physical)no abstracts in English
